0 前言
Molclus是免费灵活易用的团簇构型与分子构象搜索程序,如果不熟悉的话,请先看官网http://www.keinsci.com/research/molclus.html和《使用molclus程序做团簇构型搜索和分子构象搜索》(http://bbs.keinsci.com/thread-577-1-1.html)中的介绍。Molclus做构型/构象搜索需要提供含有初始结构信息的多帧xyz文件(通常叫traj.xyz),如笔者写的多篇Molclus教程所示,这样的文件可以通过其它程序跑分子动力学(MD)程序生成,也可以通过Molclus自带的genmer或gentor生成。本文的目的是通过一个实例,演示用Molclus基于xtb程序通过GFN-xTB方法(一种半经验形式的DFT)跑出的分子动力学轨迹做高精度分子构象搜索。
如果你还对xtb一无所知,看《将Gaussian与Grimme的xtb程序联用搜索过渡态、产生IRC、做振动分析》(http://sobereva.com/421)中的相关介绍。安装方式在里面都写明了。
本文的做法相对于以其它方式产生traj.xyz文件有以下好处:
(1)相对于用经典力学程序跑MD,用xtb跑MD极其简单,不需要经过准备拓扑文件、补参数之类麻烦的过程,初学者可以轻松上手。而相对于用量子化学程序跑从头算动力学,用xtb基于GFN-xTB理论来跑能快几个数量级,用比较烂的机子对常见大小的有机分子也能跑出足够长的轨迹。
(2)虽然Molclus的gentor也可以利用系统式扫描的方式产生含有分子各种初始构象的文件,但对于含有构象可变的环状区域的体系(比如环己烷有椅式和扭船式两种极小点),gentor没法考虑环的构象。而跑动力学的话,环状区域的各种构象是可以自然而然地被采样的。
(3)对于可旋转的键非常多的体系(比如明显超过10个键),靠gentor的话要考虑的初始构象过多。而靠动力学的话,可以通过跑的时间长度来控制最终能找到能量最低构象的概率。在参数合适的前提下,跑的时间越长,最终能找到能量最低构象的概率就越高。
注:跑动力学产生traj.xyz的缺点是如果跑的时间不够长或条件不适合,最终得到的能量最低构象会对跑动力学的初始构象有依赖性,而且有随机性(相同流程两次跑出来的结果可能不同),因为此时不像gentor系统式扫描那样可以基本保证势能面所有势阱都能被采样到。
本文下面的例子涉及的文件都可以在这里下载:http://sobereva.com/attach/532/file.rar
本文例子使用的各个程序和版本:Molclus 1.9.2,xtb 6.3pre2、Gaussian16 A.03、ORCA 4.2.1。机子是Intel 2*XEON E5-2696v3(36核),系统是CentOS 7.3 64bit。
2 被考察的体系:瑞德西韦
本文的例子是对瑞德西韦(Remdesivir)在水环境中做构象搜索,确定能量最低的和最低的一批构象。此分子是美国吉利德公司研制的一种用于抗埃博拉病毒的药物,但效果一般,在2020年1月26日被美国医生用于治疗一个患有发源于武汉的肺炎(2019-nCoV)病毒的感染者,发现效果极佳,之后吉利德公司向中国超低价慷慨援助了大量瑞德西韦。此分子的结构如下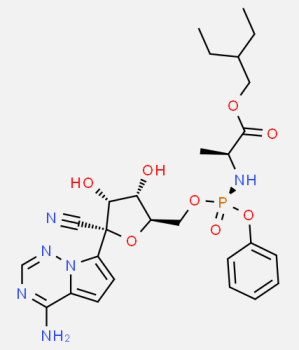
获得此体系三维结构很简单。去chemspider(https://www.chemspider.com)搜Remdesivir,然后在结构图下面点3D按钮,再点Save按钮就得到了.mol文件。里面是基于二维结构经验生成的三维结构。
之后用xtb做计算时需要基于xyz格式的文件才行。把mol文件转成xyz文件办法很多,比如Multiwfn就可以。Multiwfn可以在其官网http://sobereva.com/multiwfn免费下载。启动Multiwfn,载入本文文件包里的Remdesivir.mol,然后输入100,再选子功能2,再选择导出xyz文件的选项,然后输入导出的文件名Remdesivir.xyz,文件就马上输出在了当前目录下。
3 计算流程说明
对瑞德西韦这个体系做构象搜索流程如下。虽然过程看起来有点多,但这是保证结果高度可靠的前提下耗时最低的流程。有个几十核的像样服务器的话不超过一天时间就能搞完。
(1)用xtb在GFN0-xTB下跑动力学
解释:GFN0-xTB是GFN-xTB系列中非常便宜的一个版本,由于形式简单、不需要SCF迭代,而且xtb代码效率极高,因此比一般的半经验方法耗时还低得多得多,跑小分子的动力学极其适合。虽然GFN0-xTB计算的能量并不算太准确,但用于动力学采样的目的足够可靠了。模拟温度建议用比常温明显要高的400K,使得模拟过程构象变化较快,在有限的模拟时间内能更充分采样。模拟时间设100 ps,对于这样的小分子而言长度基本够了(但有条件的话建议跑几百ps)。目前GFN0-xTB还没有官方宣称支持溶剂模型(但估计早晚会),因此动力学过程中忽略掉溶剂效应。虽然这会导致能量、动力学轨迹和实际溶剂环境中不符,但这完全没关系,单纯对于势能面采样而言,真空下做较高温度的动力学就已经能达到目的了。
(2)通过Molclus调用xtb,在GFN0-xTB下对动力学的每一帧进行批量优化
解释:过程(1)得到的动力学轨迹通常含有几千帧,我们需要对每一帧进行几何优化然后对比能量。但这么多帧都直接用像样的方法做几何优化是极度困难的。实际上有很多帧在优化之后会收敛到相同的结构,因此可以先用极快的GFN0-xTB对所有帧进行优化,之后用Molclus里的isostat去重后,也就剩几百个结构。
(3)通过Molclus调用xtb,对(2)产生的每一个结构用GFN2-xTB在溶剂模型下进行批量优化
解释:目前还有几百个结构,都直接用DFT优化还是太昂贵。因此先用比GFN0-xTB更可靠但也更贵的GFN2-xTB进行批量优化。虽然其耗时是GFN0-xTB的2~20倍,但仍低于DFT好几个数量级。此时应当考虑GBSA溶剂模型,这使得优化后得到的能量也是有意义的。用isostat对优化后的结果进行排序,选择能量最低的一批保留下来。
(4)通过Molclus调用Gaussian,对(3)筛选出来的结构用B3LYP-D3(BJ)/6-31G*在真空下做优化和振动分析得到较可靠的结构和自由能热校正量,再调用ORCA对每个结构在PWPB95-D3(BJ)/def2-QZVPP结合SMD模型表现的水环境下算高精度单点能,二者相加得到水环境下的高精度自由能
解释:现在的候选结构已经比较少了,可以用DFT做优化和振动分析、再用高精度级别算单点能了。做振动分析一方面是为了获得自由能的热校正量,一方面是检验优化出的结构有无虚频,有虚频的结构肯定不能要。B3LYP-D3(BJ)/6-31G*是做几何优化和振动分析的最低可接受级别了,虽然看起来略low,但对于优化和振动分析目的足够了。这个过程不必考虑溶剂模型,因为哪怕是极性溶剂,对一般体系的几何结构影响也非常微小,而且用溶剂模型后往往几何优化收敛所需步数会增加、收敛更困难一些,耗时也高一点。对于局部呈现显著离子性的体系则优化和振动分析时也要考虑溶剂模型,否则结果可能显著和实际情况不符。之所以高精度单点部分改用ORCA程序,是因为ORCA做PWPB95-D3(BJ)/def2-QZVPP这样双杂化泛函结合很大基组的计算,结果又准确而耗时也不是特别高,而Gaussian里双杂化泛函结合这样的基组算当前体系绝对算不动。用PWPB95-D3(BJ)的理由我在此文写明了:《简谈量子化学计算中DFT泛函的选择》(http://sobereva.com/272)。
PS 1:在ORCA里用DLPNO-CCSD(T)算单点可以得到更准确的结果,但对于当前这个77个原子的不小体系来说,其结合较好带弥散函数的3-zeta基组(如jun-cc-pVTZ)或者def-QZVPP基组时的耗时和内存、硬盘消耗都很高,所以不用于本文例子。但如果你的体系明显小于本文的例子,或者对本文大小的体系你能忍较长的耗时的话也可以用DLPNO-CCSD(T)。若用NormalPNO版本的DLPNO-CCSD(T)结合cc-pVTZ的话,在笔者的Intel 36核机子上算瑞德西韦的单点的耗时是PWPB95-D3(BJ)/def2-QZVPP的两倍,为两个半小时。但这样不带弥散函数的3-zeta基组对于描述分子内弱相互作用还是差点意思,所以不太推荐用。
PS 2:虽然用Spartan、Macromodel、GMMX等一些傻瓜式的构象搜索程序基于分子力场做构象搜索仿佛看起来又简单又快,但由于主流分子力场精度太low,结果可靠性极低,根本没多大实际意义,而且也不像本文的做法那样可以用于包含几乎所有元素的任意类型的体系、能合理地考虑溶剂效应。在Int. J. Quantum Chem., 118, e25512 (2018)文中通过大规模测试发现哪怕是MMFF94这样的高精度有机分子力场用于算有机体系,算的构象相对能量也颇不理想,对能量最低一批结构的排序也没多大可信度,远不如PM7等主流半经验方法。虽然Grimme也搞了个名为crest的利用xtb跑metadynamics的构象搜索工具,但从实效上看并不比本文的做法更有任何优势,过程也不像本文做法那么透明和可控,且最终也还是得借助Molclus用更高级别来refine才行,所以我不认为crest有多大实际使用价值。
4 第一步:用xtb在GFN0-xTB下做动力学模拟
创建一个含有xtb的MD任务设置的文件md.inp,内容如下,双斜杠后面的是注释。
$md
temp= 400 //温度(K)
time= 100.0 //模拟的总时间(ps)
dump= 50.0 //每多少fs往轨迹文件里写入一次
step= 1.0 //步长(fs)
hmass=1 //氢原子的质量是实际的多少倍
shake=1 //将与氢有关的化学键距离都用SHAKE算法约束住
$end
更多的选项介绍看在线手册相应页面https://xtb-docs.readthedocs.io/en/latest/md.html。这里之所以把hmass明确设为1,是因为默认设置下hmass是4,即氢原子质量是实际的四倍,目的是减缓氢的运动,从而能用更大步长(即跑同样的时间可以用更少的步数),但这终究会给动力学行为带来人为虚假性,一般做MD也很少这么干。步长用的是1 fs,这是很稳妥的。其实由于用了shake=1,步长设2 fs也不是不行,但考虑到当前是较高的温度下模拟,原子运动较剧烈,为了确保动力学稳定性,还是用了保守的1 fs。保存间隔设50 fs还是比较合适的,设得太小的话相邻的帧结构太相似,之后要处理的不必要的结构太多;而设得太大的话,MD过程中的很多有意义的结构又会漏过去。模拟总长是100 ps = 100000 fs,因此最终得到的轨迹包含100000/50=2000帧。
确保xtb已正常安装了,然后运行xtb Remdesivir.xyz –input md.inp –omd –gfn 0。其中–omd告诉程序先做几何优化,然后再跑MD。做几何优化是为了避免由于初始结构太烂,导致一开始某些原子受到过大的斥力使其速度太大而令动力学崩溃。–gfn 0代表用GFN0-xTB。
MD阶段一开始会显示模拟的速度:
est. speed in wall clock h for 100 ps : 0.68
即模拟每100 ps耗时0.68小时,当前我们要跑的总长度也正好是100 ps,因此预计总耗时为60*0.68=41分钟。在笔者的Intel 36核服务器上,此任务最后花了42分钟跑完。计算过程中当前跑到第多少帧了会在屏幕上直接提示,并且不断往当前目录下的xtb.trj文件中写入结构。为了监控模拟的合理性,在任务跑的中途就可以把xtb.trj拷出来并改名为xtb.xyz,然后拖到免费的可视化程序VMD(http://www.ks.uiuc.edu/Research/vmd/)的VMD main窗口里观看已跑出来的轨迹,如果发现明显不合理,就应该赶紧停掉。每经过1 ps模拟,就会在当前目录下产生scoord.为开头的文件记录了此时的结构。
将最终的xtb.trj改名为traj.xyz,用于下一阶段的计算。此文件对应于本文文件包里xtb_opt目录下的traj.xyz,如果看这个轨迹的话,就会发现分子不断大幅运动,因此这条轨迹的采样足够充分了(另外,跑完后当前目录下还会有mdrestart,记录了最后一帧的坐标和速度,如果感觉跑的还不够长,此文件可以用于续跑)。
5 第二步:用Molclus调用xtb在GFN0-xTB下做批量优化
去Molclus官网下载最新版本并解压。把traj.xyz放到Molclus目录下。把Molclus目录下settings.ini里的iprog设为4代表调用xtb,确保itask为0代表是做优化任务。把xtb_arg参数设为”–gfn 0 –chrg 0 –uhf 0″,代表这是中性单重态体系且用GFN0-xTB计算。之后输入./molclus命令启动当前目录下的Molclus程序可执行文件,程序就开始调用xtb优化traj.xyz里的每一帧了。在笔者的Intel 36核机子上优化每一帧结构的耗时也就2~5秒钟,总共花了111分钟。
(重要技巧:这个批量优化过程可以利用crest程序来降低一个数量级的耗时,见本文最后一节的说明)
任务结束后,优化后的每一帧的结构和能量就都存到当前目录下的isomers.xyz里了。使用isostat对isomers.xyz里的结构去重和做能量排序,即输入
./isostat
[回车] //处理当前目录下的isomers.xyz
0.5 //能量去重阈值(kcal/mol)
0.5 //结构去重阈值(埃)
[回车] //不计算Boltzmann分布比例
期间在屏幕上会看到下面的信息:
The lowest energy is -122.66129225 a.u.
Sorting clusters according to energy…
# 1 Count: 1 E= -122.661292 a.u. DGmin= 0.23 DE= 0.00 kcal/mol
# 2 Count: 3 E= -122.660372 a.u. DGmin= 0.28 DE= 0.58 kcal/mol
# 3 Count: 5 E= -122.660067 a.u. DGmin= 0.17 DE= 0.77 kcal/mol
# 4 Count: 1 E= -122.659769 a.u. DGmin= 0.33 DE= 0.96 kcal/mol
# 5 Count: 3 E= -122.659587 a.u. DGmin= 0.20 DE= 1.07 kcal/mol
# 6 Count: 1 E= -122.659178 a.u. DGmin= 0.22 DE= 1.33 kcal/mol
…略
# 268 Count: 2 E= -122.637199 a.u. DGmin= 0.08 DE= 15.12 kcal/mol
# 269 Count: 3 E= -122.637073 a.u. DGmin= 0.33 DE= 15.20 kcal/mol
# 270 Count: 4 E= -122.636938 a.u. DGmin= 0.44 DE= 15.28 kcal/mol
# 271 Count: 1 E= -122.636612 a.u. DGmin= 0.28 DE= 15.49 kcal/mol
# 272 Count: 1 E= -122.635534 a.u. DGmin= 0.33 DE= 16.16 kcal/mol
最后得到的共272个非重复结构产生在了当前目录下的cluster.xyz里。删掉之前的traj.xyz,然后把cluster.xyz改名为traj.xyz,使得这些去重后的结构作为接下来Molclus任务的输入结构。
6 第三步:用Molclus调用xtb在GFN2-xTB结合隐式水模型下做批量优化
把settings.ini里的xtb_arg参数设为”–gfn 2 –gbsa h2o –chrg 0 –uhf 0″,代表用GFN2-xTB级别结合GBSA模型表现的水环境进行计算。启动molclus来调用xtb对当前272个结构进行优化。优化每个构象耗时十几秒钟,整个任务耗时48分钟(这个批量优化过程可以利用crest程序来降低一个数量级的耗时,见本文最后一节的说明)。之后运行isostat进行去重和排序,做法同前,输出如下:
# 1 Count: 1 E= -128.462874 a.u. DGmin= 0.29 DE= 0.00 kcal/mol
# 2 Count: 2 E= -128.462743 a.u. DGmin= 0.25 DE= 0.08 kcal/mol
# 3 Count: 2 E= -128.460194 a.u. DGmin= 0.31 DE= 1.68 kcal/mol
# 4 Count: 1 E= -128.459966 a.u. DGmin= 0.50 DE= 1.82 kcal/mol
# 5 Count: 1 E= -128.459373 a.u. DGmin= 0.24 DE= 2.20 kcal/mol
# 6 Count: 1 E= -128.458599 a.u. DGmin= 0.24 DE= 2.68 kcal/mol
# 7 Count: 2 E= -128.457983 a.u. DGmin= 0.22 DE= 3.07 kcal/mol
# 8 Count: 2 E= -128.457949 a.u. DGmin= 0.17 DE= 3.09 kcal/mol
# 9 Count: 3 E= -128.457877 a.u. DGmin= 0.18 DE= 3.14 kcal/mol
# 10 Count: 9 E= -128.457797 a.u. DGmin= 0.36 DE= 3.19 kcal/mol
…略
目前非重复的一共有148个结构。虽然当前的能量已经有一定实际实际意义了,能量高低次序已经能一定程度体现不同构象的能量高低了,但这个体系柔性高,能量最低的一批构象的能量肯定很相近,靠GFN2-xTB的精度还是明显不足以可靠地区分能量差很小的不同构象的能量相对高低。而且xtb用的GBSA溶剂模型也远不如量子化学程序用的PCM等溶剂模型可靠。而且当前也只是考虑的电子能量,没考虑自由能。因此,之后还需要用更好的级别做几何优化、振动分析、单点计算来得到准确的溶液下的自由能,这就是接下来要做的事。
GFN2-xTB在GBSA模型下给出的相对能量虽然不算很准,但也定性正确,分辨几个kcal/mol的差异还是没问题的。我们这里就保留相对能量<=3 kcal/mol的6个结构用于下一步的精炼(这个标准是随意设的。假设对于你的情况<=3 kcal/mol的都有50个结构,还是觉得太多,那可以比如只保留能量最低的20个。保留多少要结合实际计算条件、对精度要求等方面自行权衡。为了稳妥起见,条件允许的话我建议保留所有<=4或<=5 kcal/mol的构象)。
还是将traj.xyz删掉,把cluster.xyz改名为traj.xyz。
7 第四步:用Molclus调用Gaussian和ORCA得到水环境下的高精度自由能
把settings.ini里的iprog设为1代表调用Gaussian。ngeom设为6代表只处理traj.xyz里的前6个结构(即只考虑上一步得到的能量最低的6个结构,因为isostat给出的cluster.xyz是按照能量由低到高排序的)。itask设为3代表做优化+振动分析来得到自由能。把gaussian_path和orca_path分别设为调用机子里Gaussian和ORCA的实际命令。另外建议把ibkout设为1代表把算每个体系的输出文件都进行备份,便于之后必要时进行检查。
把molclus目录下Gaussian模板文件template.gjf里的关键词设为B3LYP/6-31G* em=GD3BJ opt freq。代表在B3LYP-D3(BJ)/6-31G*下做优化和振动分析。PS:如果你是Gaussian 16的用户,建议加上int=fine,能节约很多时间。默认的int=ultrafine对于B3LYP-D3(BJ)来说太浪费了。
在molclus目录下创建一个叫template_SP.inp的ORCA输入文件的模板文件,内容如下,对应在RI-PWPB95-D3(BJ)/def2-QZVPP结合SMD模型表现的水环境下做高精度单点计算。如果对ORCA的这些关键词、计算级别不熟悉,不知道为什么这么写的话,强烈建议看《详谈Multiwfn产生ORCA量子化学程序的输入文件的功能》(http://sobereva.com/490)。
! PWPB95 D3 def2-QZVPP def2/J def2-QZVPP/C RIJCOSX grid4 gridx4 tightSCF noautostart miniprint nopop
%maxcore 6000
%pal nprocs 36 end
%cpcm
smd true
SMDsolvent “water”
end
* xyz 0 1
[GEOMETRY]
*
在Molclus每次调用Gaussian做完优化和振动分析任务后,当程序发现当前目录下有template_SP.inp文件时,就会自动调用ORCA做单点计算,并将所得的高精度单点能自动加到振动分析得到的自由能热校正量上作为此结构的最终能量,这也正对应于溶剂下的自由能(严格来说还需要加上1.89 kcal/mol标准态转换能量后才是真正意义的溶剂下的自由能,详见http://sobereva.com/327。但是否考虑这个不影响构象间相对能量)。
现在执行./molclus |tee out.txt,这代表运行molclus的时候也把输出信息写入当前目录下的out.txt里便于之后检查。
计算过程中,调用Gaussian计算时,正在运行的Gaussian任务的输出文件名是gau.out,故可以通过tail -f gau.out命令监控计算进程,即让gau.out里新输出的信息不断显示在屏幕上。当进入调用ORCA算单点能的时候,也可以通过tail -f orcaSP.out监控计算进程。在笔者的Intel 36核机子下,对每个结构Gaussian做opt freq任务耗时都是一个小时多一点,ORCA算每个高精度单点任务也是耗时一个小时多一点,因此每个结构共花两个多小时,6个结构一共花了15个小时。
每个结构振动分析完成后Molclus会在屏幕上提示有没有虚频,在isomers.xyz文件里每个结构的Nimag =字样后面也可以看到虚频数目。当前的计算得到的每个结构都没有虚频,所以最终结果是有意义的。如果有的有虚频,应当加上有助于消除虚频的关键词重新计算,见《Gaussian中几何优化收敛后Freq时出现NO或虚频的原因和解决方法》(http://sobereva.com/278)。
算完后,还是用isostat处理得到的isomers.xyz,输出信息如下。可见这次有两个结构经过优化后收敛到了相同结构,故最终剩下5个。如前所述,当前显示的能量对应的直接就是溶剂环境下的自由能。
Energy of isomer 1 : -2320.81823800 a.u., new cluster
Energy of isomer 2 : -2320.81480600 a.u., new cluster
Energy of isomer 3 : -2320.80966500 a.u., new cluster
Energy of isomer 4 : -2320.81852000 a.u., new cluster
Energy of isomer 5 : -2320.81485300 a.u., attributed to cluster 2
Energy of isomer 6 : -2320.80904200 a.u., new cluster
The lowest energy is -2320.81852000 a.u.
Sorting clusters according to energy…
# 1 Count: 1 E= -2320.818520 a.u. DGmin= 1.25 DE= 0.00 kcal/mol
# 2 Count: 1 E= -2320.818238 a.u. DGmin= 1.01 DE= 0.18 kcal/mol
# 3 Count: 2 E= -2320.814853 a.u. DGmin= 1.01 DE= 2.30 kcal/mol
# 4 Count: 1 E= -2320.809665 a.u. DGmin= 1.10 DE= 5.56 kcal/mol
# 5 Count: 1 E= -2320.809042 a.u. DGmin= 1.10 DE= 5.95 kcal/mol
可见最低的两个构象能量相当接近,都不可忽略。虽然当前用的计算自由能的做法已经很好了,但由于SMD算中性体系的溶解自由能通常都有零点几kcal/mol误差(这还只是对于小体系而言),而且即便RI-PWPB95-D3(BJ)/def2-QZVPP给出的构象相对能量差也并不能保证总能精确到零点几kcal/mol,所以当前我们应当认为我们找出来的能量最低的两个构象都算是最稳定构象,在实际水环境中出现的几率都会非常大。
接着在isostat里输入y,再输入298.15,就会得到常温下的Boltzmann分布比例,如下所示,原理详见《根据Boltzmann分布计算分子不同构象所占比例》(http://sobereva.com/165)。
# 1 Count: 1 Ratio: 56.74%
# 2 Count: 1 Ratio: 42.09%
# 3 Count: 2 Ratio: 1.17%
# 4 Count: 1 Ratio: 0.00%
# 5 Count: 1 Ratio: 0.00%
可见,常温下前两个构象共同占据绝对主导,而其它构象的出现概率就都可以忽略了。
PS:如果你要考察其它温度下的Boltzmann分布,不仅isostat里要输入相应的温度,在template.gjf里也应当加上“temperature=[温度]”关键词来让Gaussian输出的自由能热校正量对应相应的温度;或者settings.ini里用ibkchk=1,使得每个opt freq任务的chk都被保留下来,便于之后自行用freqchk工具得到其它温度下的自由能热校正量。单点能从Molclus的ibkout选项备份出来的ORCA输出文件里直接就可以读到。
下面是自由能最低的那两个构象的结构图,即对应于isostat给出的cluster.xyz的前两帧: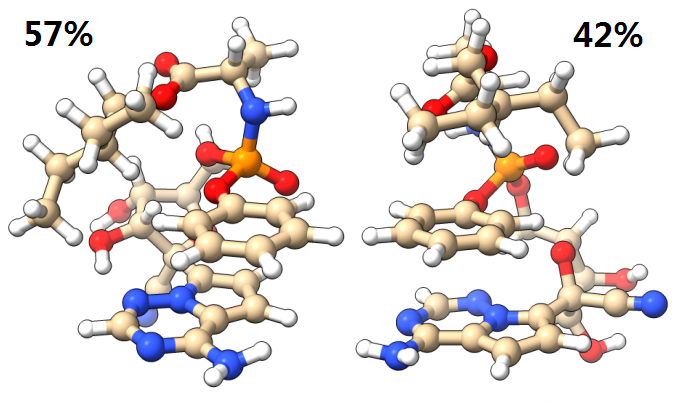
能量最低构象的旋转动画如下,分子的各个部位都能看清楚

8 弱相互作用分析
这里顺便对上面得到的两种能量最低构象考察一下分子内相互作用。Multiwfn支持的弱相互作用分析方法极多,见《Multiwfn支持的弱相互作用的分析方法概览》(http://sobereva.com/252)。为了便于构象搜索后用Multiwfn做这些分析,个人建议Molclus调用Gaussian做opt freq任务时ibkchk总是设为1从而自动保留chk文件。
这里就用《使用Multiwfn图形化研究弱相互作用》(http://sobereva.com/68)中介绍的已经很流行的NCI方法考察一下能量最低的两个构象的分子内弱相互作用,NCI图如下所示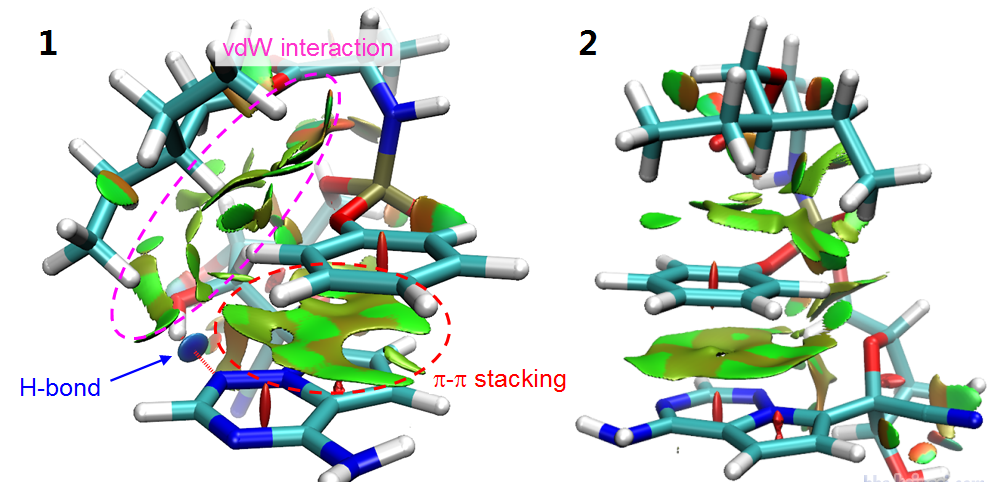
由上图可见,这两个构象主要差异在于碱基和苯基之间的相对位置以及分叉的烃链的位置。2的pi-pi堆积更强一些,因为苯环与碱基错开的程度更低,而1中分叉烃链可以与碱基与苯基同时发生范德华吸引作用,而且还有2所没有的O-H…N内氢键。因此二者都有各自的对自己构象有利的分子内相互作用。如果你去看其它构象的话,就会发现分子内相互作用明显不如1和2这么充分和有利,因此能量明显更高。
9 附加说明
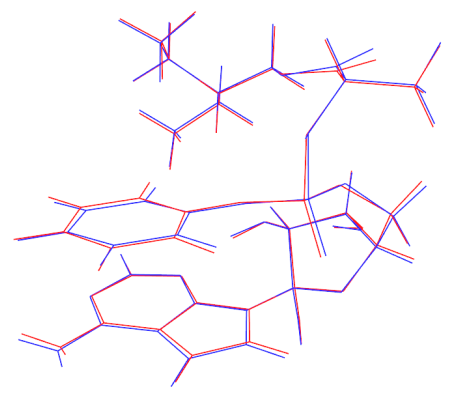
本文介绍的构象搜索方法普适性极强,几乎什么体系都可以用,各类分子、分子团簇、原子团簇皆可。只不过要注意GFN-xTB系列方法目前只能用于<=86号元素,因此碰上锕系配合物等没法用。并且原始的6-31G*只支持前四周期元素,如果涉及更靠后的元素,最简单的办法是把基组改为def2-SVP,耗时只高出一点,对前六周期元素都有定义。本文的构象搜索流程是否一定能找到能量最低的构象很大程度上取决于体系特征、xtb做动力学跑的时间长短和温度,以及GFN2-xTB优化后保留最低的多少个构象用于之后的DFT计算。显然体系可旋转的键越少、动力学跑的越长、保留的构象越多,则最终能找到能量最低构象的成功几率就越高。动力学用较高温度也有助于翻越较高势垒,从而更全面地采样,但温度过高可能会导致动力学不稳定、出现不该有的异构化结构、在能量较低势能面区域采样不充分等问题。对于瑞德西韦这个体系我们所找出的两个能量最低构象,从几何结构和分子内弱相互作用模式来看都极其合理,从直觉上也想象不出还有什么其它构象能有更低的能量,因此可以断定我们成功找到了真实的能量最低构象。几何优化和振动分析调用ORCA来做也不是不行,特别是如果你没有Gaussian的话这是首选。但ORCA的优化算法不如Gaussian稳健,而且振动分析的效率低,所以虽然ORCA利用RI技术可以令每一步优化耗时较低,但总的来说除非体系特别大(如一二百个原子),否则仍然更建议用Gaussian。ORCA支持的B97-3c方法做优化又好又快(大概率好于B3LYP-D3(BJ)/6-31G*,而耗时更低),但是对于笔者撰文时的最新的4.2.1版,B97-3c算的Hessian似乎有点bug,容易出现小虚频,虽然用numfreq数值频率可以解决,但此时耗时太高,故应当慎用。有人可能会觉得6-31G*对于弱相互作用明显影响分子构象的柔性大体系的优化可能太小了。实际上用这个就已经够了。为体现这一点,笔者对前面得到的能量最低构象在6-311G**下做了进一步优化,原先的6-31G*的结构和新得到的6-311G**的结构经过叠合后的图像如下所示,红色是6-31G*的,蓝色是6-311G**的,可见没什么明显区别。内氢键键长、pi-pi堆积的平面间距也基本没变,而用6-311G**优化此体系耗时比用6-31G*高4、5倍。不过,如果当前体系只有几十原子,你的计算资源又很富裕,为了避免跟某些较真的审稿人argue,也可以用6-311G**(而再继续提升基组就绝对没有丝毫意义了)。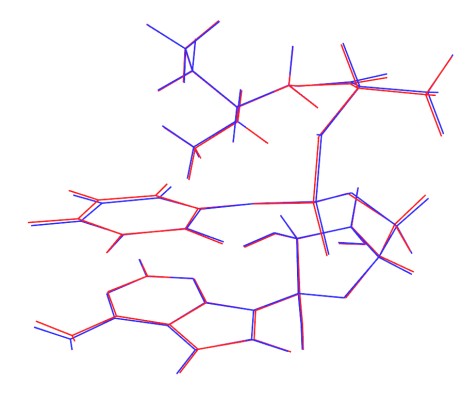
本例计算时在DFT做优化过程中没有考虑溶剂模型,不用担心这会造成明显问题。对前面找出来的能量最低结构,笔者之后又在IEFPCM模型表现的水环境下在相同级别做了优化。下图是气相下(红线)和水模型下(蓝线)优化后的叠合图,可见结构差异基本可以忽略。
不过,如果希望更严谨一些,避免审稿人对计算流程吐槽什么的,并且能接受更高一些的耗时(高几分之一),Gaussian做DFT优化的步骤也可以用Gaussian默认的IEFPCM隐式溶剂模型。注意此时不推荐用SMD模型,因为对于优化和振动分析这种目的不是算能量的任务它不会比IEFPCM更好,而且有时会出现收敛困难以及造成小虚频。如果是用ORCA来做优化和振动分析,更不建议这个阶段用溶剂模型,因为ORCA的溶剂模型的代码不如Gaussian成熟稳健,哪怕不用SMD而用CPCM,ORCA对柔性大体系优化出来的结构也容易有小虚频。而且ORCA里对大体系开溶剂模型导致耗时的增加明显高于Gaussian。
在GFN2-xTB优化后用DFT再做优化和振动分析是绝对必要的,不要图省事直接就用GFN2-xTB优化的结构算高精度单点。对于瑞德西韦这种柔性大分子,若基于GFN2-xTB的结构再做DFT优化,通常会看到结构还有显著的变化,即第一帧和最后一帧肉眼都能看出不同。为了说明这一点,对前面我们得到的能量最低结构(蓝线)我用GFN2-xTB在真空下又做了一下优化(红线),经过叠合后的图如下所示,可见从同一个初猜结构开始,GFN2-xTB的极小点结构和DFT极小点结构相差非常明显,这样的差异是绝对不能接受的(实际上这个构象还算是相差相对较小的,本文研究过程中还发现DFT和GFN2-xTB优化出来的某些构象里烷烃链那部分位置差得老远)。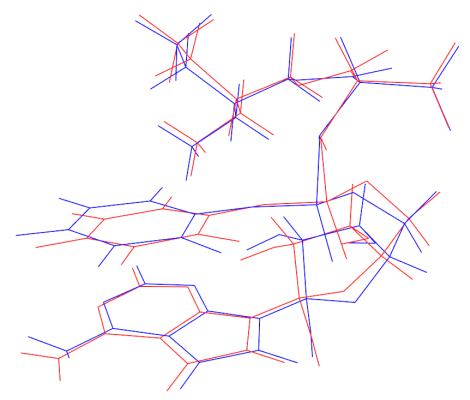
笔者在《谈谈谐振频率校正因子》(http://sobereva.com/221)中介绍过频率校正因子,原理上来说计算自由能热校正量时考虑校正因子有益,但从对实际构象能量差的影响程度来看考虑不考虑都无所谓,所以本文就没考虑。要考虑的话可以在opt freq任务对应的template.gjf里加上scale=关键词,就用ZPE校正因子就可以。
ORCA算高精度单点用的PWPB95-D3(BJ)/def2-QZVPP对于比当前体系显著更大的体系可能会算得很吃力,届时可以改为def2-TZVPP。但注意QZ级别才能充分发挥双杂化泛函的潜力。对于PWPB95-D3(BJ)/def2-TZVPP都很难算得动的体系,可以改为普通泛函ωB97M-V结合def2-TZVPP,参见此文的说明《详谈Multiwfn产生ORCA量子化学程序的输入文件的功能》(http://sobereva.com/490)。
本文的算溶剂下的自由能的做法是懒人的做法,即“真空下高精度单点能”和“溶解自由能”这两部分的计算直接通过一个任务“溶剂下高精度单点能”完成,这样做原理上没有用M05-2X/6-31G*专门结合SMD模型去算溶解自由能那么理想,详见《谈谈隐式溶剂模型下溶解自由能和体系自由能的计算》(http://sobereva.com/327)。不过这也不是什么明显问题,更何况构象能量求差的时候误差还能抵消很多。
本文的构象搜索的全套流程完全可以写成shell脚本自动完成,因为涉及的所有操作环节都能简单地通过命令行完成,Molclus也是通过命令行运行的。感兴趣的读者请自行完成这样的脚本,并不复杂。
有本文读者问及用crest做metadynamics如何,前文里已经说了,有这个没有什么实际好处。笔者也实际试了一下,非常令人失望:
crest结合GFN2-xTB真空下搜索,两个半小时结束,给出的能量最低的至少前50个结构里没有任何一个和真实的能量最低结构有相似性
crest结合GFN2-xTB在GBSA表现的水溶剂下搜索,两个小时结束,给出的能量最低的至少前50个结构里没有任何一个和真实的能量最低结构有相似性
crest结合GFN0-xTB真空下做搜索,45分钟结束,所有给出的516个结构没有任何一个结构和真实的能量最低结构有相似性
可见对于当前体系大失败。crest的原理看似sophisticated,实际表现还没高温MD效果好。
技巧:利用crest程序节约xtb批量优化大量结构阶段的耗时
在前面文章中我们看到,Molclus结合xtb可以快速对xyz文件里记录的大批量结构进行快速优化。实际上这个阶段还可以明显节约时间,也就是利用crest程序。crest可执行程序在xtb的github页面上就能下载到。Molclus调用xtb时是对traj.xyz文件里的结构一个接一个来优化的,xtb在执行时是用OMP_NUM_THREADS环境变量指定的线程数做并行计算。而crest调用xtb来优化时,会同时启动OMP_NUM_THREADS个xtb程序同时对输入的xyz里的结构进行批量优化,每个xtb都只用单线程。crest这种方式调用xtb做批量优化的总耗时明显更低。
具体来说,运行以下命令就可以在GFN0-xTB级别下用normal几何优化收敛限(这是xtb默认的收敛限)对traj.xyz里所有结构进行优化:
/sob/crest -mdopt traj.xyz -gfn0 -opt normal -niceprint
其中/sob/crest是crest可执行文件路径,-niceprint代表计算过程中显示进度条。还可以用-chrg指定体系净电荷数,-uhf指定自旋,-g [溶剂名]指定溶剂模型。
计算完毕后在当前目录下得到了crest_ensemble.xyz文件,记录了优化后每一帧的结构和能量。从Molclus 1.9.3版开始,自带的isostat工具也能处理这个文件,当发现文件名里面有crest字样的时候就会自动以crest_ensemble.xyz文件的格式来读取内容,然后进行去重、排序并得到cluster.xyz。
用此做法对本文例子里高温MD跑完后的2000个结构用此方法优化,在笔者的Intel 36核机子上只花了11分钟,比起用Molclus调用xtb优化的时间节约了一个数量级。类似地,之后用GFN2-xTB在水模型下批量优化的那个阶段也可以借用crest来显著降低耗时。