Computational modelling has now become a routine procedure in all branches of chemistry. From modelling crystals to organic reaction mechanisms, it is being used to predict data,understand it, or to explain it.
However, the learning curve for this field is quite steep. Very few softwares are user-friendly, and most do not have a graphical interface. Efficient softwares like Gaussian and Spartan exist, but they are both commercial. When I first started to learn this, I found this very problematic, as I wanted to try it before I committed to it. While there are books that explain the theory of computational chemistry in detail, there is a distinct lack of books that explain how to use the software: the technical side of the field.
In this series of blogs, I will try to give a brief, non-technical introduction to computational modelling from what I have learnt (and I am in no way an expert in this). I will also attempt to explain some of the theory behind modelling as we go along, but it would not be very detailed (and you can always skip them if you want to!). The modelling will be done mainly with GAMESS, and visualisation with Avogadro and wxMacMolPlt. Some other softwares will also be occasionally used. These are all freely obtainable online.
Installing GAMESS
If you are on Windows or Mac, then your best bet is to download the precompiled installer from the GAMESS website. Install the software, then follow the instructions on the pdf in the installation folder (you have to install MS-MPI for windows), and it should work without any problem. If you are on linux, then you can use this or this guide for help.
Installing Avogadro
Download and install Avogadro from avogadro.cc.
Installing wxMacMolPlt
Go to this website to install wxMacMolPlt.
Drawing the structure
Draw the structure in Avogadro using the draw tool:
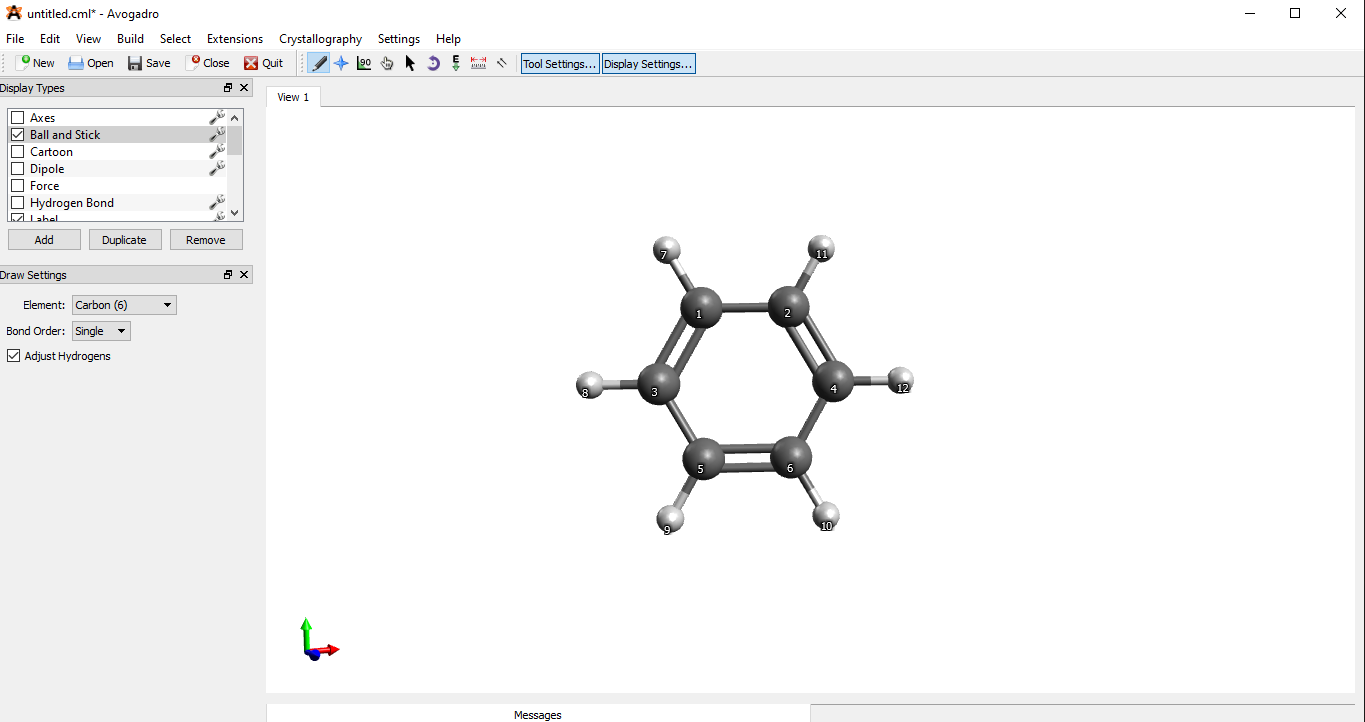
First draw the structure, and then use the Auto Optimization Tool (the E symbol) to optimize the drawn structure — this will fix stretched bonds, odd angles etc. This is going to be our starting structure i.e. guess. (Remember! auto-optimization is based on force-fields which do not always give correct results, especially for inorganic compounds). The choice of force-field is important: MMFF94s is good for organic molecules, UFF can be used with all elements, but might give very wrong results.
Creating input file for GAMESS
GAMESS requires an input file (.inp) that contains the 3D coordinates of the molecule, and instructions on what calculation to run. Generating input with Avogadro is easy, but we have to edit it afterwards. Use Extensions > GAMESS > Input generator. Choose Equilibrium geometry in ‘Calculate section’ and choose B3LYP and 6–31(d) in the ‘With’ section. Leave the rest as they are.

Then select ‘Generate…’ and save the .inp file.
Editing the input
Now we have to look at the input file. Now, GAMESS input files have group keywords each of which control a particular aspect of the calculation. For example $CONTRL is the main control which decides which type of calculation has to be run. $BASIS chooses the basis sets, which are sets of functions used to approximate the atomic orbitals. $STATPT controls the optimization process. Finally, $DATA provides the coordinates of the atoms constituting the molecule.
A bit of theory now — B3LYP is a density functional, which gives good results at low computational cost (however, most density functionals have empirical parameters in them, so they should not be used blindly). 6–31G(d) is also a good basis set, even if it is a bit old.
Now, some parts of the input file have to be edited (open the file with notepad or any other text editor). Change OPTTOL=0.0001 to OPTTOL=0.0004 (tolerance for the optimization, lower means tighter optimization, but 0.0004 is okay for most purposes). Also, change NSTEP to 100 (20 optimization step is not enough). Also, create two new lines and write $ZMAT DLC=.t. AUTO=.t. $END, and $SYSTEM MWORDS=4 $END. Then, add NZVAR=33 in $CONTRL. Every line begins with a blankspace, and keywords must also be separated by a space, otherwise, the program will throw an error.
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 #END
$CONTRL SCFTYP=RHF RUNTYP=OPTIMIZE DFTTYP=B3LYP NZVAR=33 $END
$STATPT OPTTOL=0.0004 NSTEP=100 $END
$ZMAT DLC=.t. AUTO=.t. $END
$SYSTEM MWORDS=4 $ENDRunning the calculation
There are multiple ways to run the calculation. The way I run it is that I place the file in the ‘inputs’ folder in the place where GAMESS is installed. And then I hit double-click-run.bat — this runs all input files in the folder, and places the output files in the folder ‘outputs’.
This calculation should not take more than 15 minutes on most PCs. Open the output file (.log). At the end, it should say ‘execution of gamess terminated normally’ — that means the program ran successfully. Also, use the find tool to look for a line that says ‘equilibrium geometry located’. If these two things are there, that means your calculation has run successfully.
Visualising molecular orbitals
To see the molecular orbitals, open the output file with Avogadro. You should see the benzene ring, and a list of molecular orbitals (MOs) on the panel to the right. Avogadro automatically renders 5 MO’s below the highest occupied MO (HOMO) and 5 MOs above the lowest unoccupied MO (LUMO). To see other orbitals, click on them, and press ‘Render’.
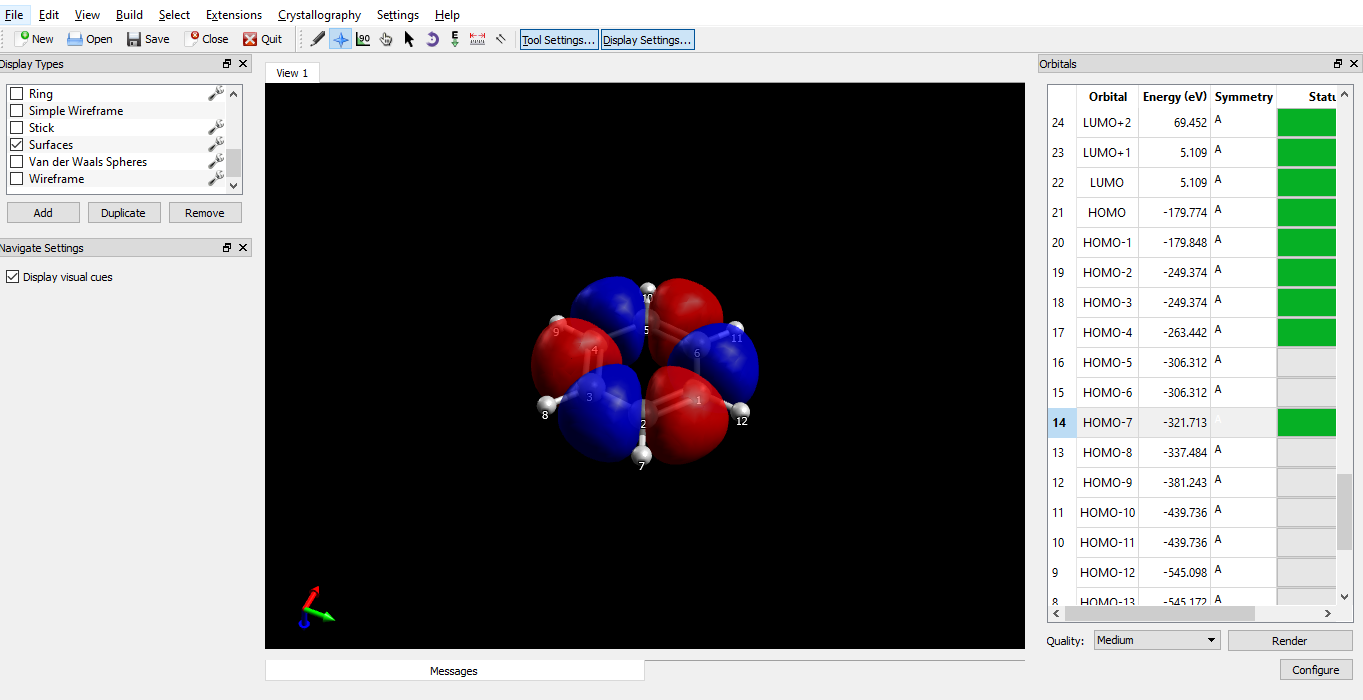
You can change the quality, but higher quality takes longer time to render. You can also use ‘Configure’ to change the isosurface value. The orbital surfaces are drawn so that on every point on that surface, the the value of the wavefunction (Ψ) is constant. This constant is determined by the isosurface value. Reducing the isosurface value makes the orbitals large and vice versa. There is no fixed rule for what the value should be, but I have used 0.03 here.
On the left side, in the ‘Display types’ panel, if you click on the spanner button next to ‘Surfaces’, there are options to change the colours of the orbitals and their opacity.
And that’s it! You can visualise the orbitals of small organic molecules following this method.
GAMESS is developed and maintained by the Gordon research group in Iowa State University: https://www.msg.chem.iastate.edu/gamess/